Mohammad A Ghattas1,
Nermin A Eissa1,
Sanaa K Bardaweel2,
Abdallah Abu Mellal1,
Noor Atatreh1 ![]()
For correspondence:- Noor Atatreh Email: noor.atatreh@aau.ac.ae Tel:+97137024888
Received: 19 August 2016 Accepted: 12 January 2017 Published: 27 February 2017
Citation: Ghattas MA, Eissa NA, Bardaweel SK, Mellal AA, Atatreh N. Computer-aided discovery of antimicrobial agents as potential enoyl acyl carrier protein reductase inhibitors. Trop J Pharm Res 2017; 16(2):397-405 doi: 10.4314/tjpr.v16i2.19
© 2017 The authors.
This is an Open Access article that uses a funding model which does not charge readers or their institutions for access and distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0) and the Budapest Open Access Initiative (http://www.budapestopenaccessinitiative.org/read), which permit unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited..
Purpose: To perform a virtual screening for a set of drug-like ligand library against the Staphylococcus aureus enoyl acyl carrier protein reductase, saFabI.
Methods: The virtual screening was conducted based on a previously validated pharmacophore-constrained docking. Consequently, the top list obtained was filtered using visual inspection where forty compounds were selected for experimental testing using disk-diffusion test and broth dilution method. The hits obtained were checked for their toxicity against human fibroblasts cell lines.
Results: Three compounds were active against Staphylococcus aureus and other tested gram-positive bacteria. However, no significant inhibitory activity (p < 0.05) was detected against Escherichia coli or Candida albicans. The minimum inhibitory concentration (MIC) values for the most active compounds were identified using the broth dilution method; all of them exhibited inhibitory activity within micromolar range. The docking results showed that the hits obtained exhibited a small size with a nice binding mode to saFabI enzyme, forming the important interactions with the key residues. Furthermore, the best three hits demonstrated good safety profile as they did not show any significant toxicity against human fibroblast cell line.
Conclusion: Overall, the newly discovered hits can act as a good starting point in the future for the development of safe and potent antibacterial agents
Introduction
Enoyl acyl carrier protein reductases (ENRs) are essential enzymes for microorganisms’ survival. These enzymes play an important role in the elongation process of synthesized fatty acids [1]. In the final step of fatty acids elongation pathway, ENRs reduce enoyl-thioester to an acyl moiety; this reduction process requires a hydride source which is provided by a bound NADH or NADPH [2]. This particular step in the fatty acid elongation pathway is shared amongst many organisms, but with significant variation in catalysing enzymes’ structure and organisation [3]. In bacteria, monofunctional proteins catalyses each reaction separately (i.e. FAS II), while in human a large multifunctional protein (i.e. FAS I) is responsible for all enzymatic activities involved in the fatty acids elongation pathway. Therefore, bacterial ENRs are believed to be good specific targets for antibacterial agents development, as they will not affect the human FAS I protein [2,3].
The Staphylococcus aureus FabI (saFabI) is a short chain dehydrogenase reductase ENR [3]. In general, ENRs have a conserved triad in its catalytic sites: Tyrosine, lysine and phenylalanine or tyrosine. Whereas the tyrosine residue has a direct role in the reduction catalysis by acting as a proton donor, lysine’s role is limited to stabilizing the binding of the cofactor [4]. The U-shaped conformation of the fatty acyl substrate is guided by the third residue which is located close to the nicotinamide ring of NADP [5]. Our previous protein-ligand interaction study carried on saFabI crystal structures revealed that the proton donor Tyr is essential for the binding of the co-crystallized inhibitors; a hydrogen bonding between them was seen in all examined complexes [6]. The bound NADP+ nicotinamide ring showed similar importance via making a π-π stacking interaction with the corresponding aromatic ring of the saFabI inhibitors. In the same study, it was revealed that constraining, particularly, the first key interaction during docking had a positive impact on the enrichment of the saFabI virtual screening as well as on the docking accuracy of the known saFabI inhibitors [6].
The increased resistance of bacteria to existing drugs makes the development of new agents a crucial step in combating infectious diseases. ENRs introduce themselves as potential targets for the development of new antibacterial agents [7] and employing the right drug design technique against them can eventually turn fruitful. In our current study, we employed a previously validated virtual screening approach [6] to target the saFabI-NADP+ complex with a drug-like ligand library. Consequently, the top-ranked compounds were brought in and tested for their antibacterial activity against a panel of Gram-negative and Gram-positive bacterial strains. The minimum inhibitory concentrations were identified for the most active hits. Promising hits were then biologically tested against human cell lines to determine their safety margin and specificity.
Methods
The saFabI crystal structure was obtained from the protein data bank (http://www.rcsb.org/), PDB: 4ALK [8]. All solvent molecules were eliminated prior to protein preparation. The MOE 3D protonate module [9] was employed to add hydrogen atoms to the protein structure and to give partial charges to each atom based on the MMFF94x force field [10].
A ligand library was obtained from the National Cancer Institute/USA (www.cancer.gov). The ligands were filtered based on the Veber’s rules [11] and the Lipinski’s rule of five [12]. These druglikeness filters included: molecular weight ≤ 500, hydrogen bond donor (HBD) ≤ 5, hydrogen bond acceptor (HBA) ≤ 10, logP ≤ 5, polar surface area (PSA) ≤ 140 and rotatable bonds ≤ 10. Ligands were protonated/deprotonated based on the Wash module in MOE [9] and were then assigned partial charges based on the MMFF94x force field [10].
Virtual screening docking protocol
A docking protocol, that had been previously validated by our group [6], was also employed here against the saFabI enzyme. Using Pharmacophore Elucidation [9], a pharmaco-phoric feature was created for use in virtual screening; the pharmacophoric point was assigned based on the NADP+ ribose 2-hydroxyl group (denoted as Acc2). Acc2 annotates projected locations of potential H-bond donors.
Subsequently, the co-crystallised ligand was used to define the saFabI catalytic pocket. Then, the prepared druglike ligand library was docked into the saFabI enzyme active site using the MOE-Dock program [9]. The Pharmacophore algorithm was used as a placement method in the constrained docking protocol. In the Pharmacophore algorithm, the ligand is placed using pre-defined pharmacophore and is then oriented to take the best fit [9]. Only poses that satisfy pharmacophoric features are considered for scoring. Affinity dG [9] was used to score and rank poses generated from the placement stage; the final docking output list included only the top ranked pose of each ligand. Affinity dG involves several terms for calculating contributions from hydrogen bonding, electrostatic and hydrophobic interactions.
Top 16000 docked ligands were refined via the MMFF94x force field [10] and were consequently given binding energies based on the GBVI/WSA dG scoring function. Top 2000 ligands of the refined list were clustered based on the MACCS algorithm [13] and were then visually inspected. Ligands from different chemical clusters that showed convenient binding modes and nice pocket filling were selected for in vitro testing.
As for the reference ligand docking and scoring, we found the active conformation of triclosan co-crystallized with saFabI in the protein data bank, PDB: 4ALL [8]. The whole structure was aligned on the protein structure used in this screening so that triclosan can get placed accurately inside the saFabI catalytic pocket. Afterwards, triclosan was refined via the MMFF94x force field and was then scored based on the GBVI/WSA dG scoring function.
Antimicrobial testing
The in vitro antimicrobial activity of all compounds against the Gram-positive Staphylococcus aureus ATCC25923 was determined. Compounds showing significant antimicrobial activity against S. aureus were selected for further testing against methicillin resistant Staphylococcus aureus (MRSA) ATCC33591, Bacillus subtilis ATCC6633, Gram-negative Escherichia coli ATCC25922 and the fungus Candida albicans ATCC10231. All strains were purchased from Remel Europe (Datford, UK). Disk diffusion method was used in the preliminary screening as per Clinical Laboratory Standards Institute [14], where nutrient agar medium (LABM, Lancashire, UK) was used for bacteria and Sabouraud dexrose agar medium (LABM, Lancashire, UK) was used for fungi. Sterile filter paper disks (6mm, sigma, St. Louis, MO or Whatman Number 1) were soaked in the test compounds dissolved in dimethylsulfoxide, (10mM, Sisco Research Lab., Mumbai, India), and were allowed to dry at room temperature in order to remove any residual solvent. The disks were then placed carefully on previously inoculated agar plates. The plates were incubated at 37 oC for 24 h (bacteria) and 30 oC for 48 h (fungi), and the diameter of the growth inhibition zones were measured to the nearest whole millimeter.
Broth dilution method was used to determine the minimum inhibitory concentrations (MIC) for the most active compounds NM02 and NM06 against S. aureus, MRSA and B. subtilis as per Clinical Laboratory Standards Institute [15] and literature [16,17]. Tested compounds were dissolved in dimethylsulfoxide where two folds dilutions of the solution were prepared and then were diluted in culture medium (Nutrient broth, LABM, Lancashire, UK) to obtain a final concentration range of 0.56 – 143 µg/mL. The dimethylsulfoxide final concentration never exceeded 1 % v/v. Each test tube was inoculated with the microorganism suspensions at 1 X 106 CFU/mL (colony forming unit/ml) concentrations. The MICs were determined after incubation at 37 oC for 24 h (bacteria). Tubes consisting of growth media with 1 % v/v dimethylsulfoxide were used as controls. The antibacterial agent; chloramphenicol (Sigma, St. Louis, MO); and the antifungal agent; ketoconazole (Bio-rad, Marnes-la-Coquette, France); were used as references. All experiments were repeated two times. The MIC values were determined as the lowest concentrations of compound that completely inhibited the visible growth of each strain as detected by unaided eye.
Cytotoxicity test
Fibroblasts were cultivated at 37 °C in Dulbecco’s modified Eagles medium (DMEM, Biochrom, Berlin, Germany) and 10 % fetal calf serum (FCS), (Gibco, Paisley, U.K.) containing penicillin/streptomycin (Sigma, St. Louis, MO) and amphotericin B (Sigma, St. Louis, MO). Cells were plated in 96-well tissue culture plates. Cells from passage number 9-15 were used. For the cytotoxicity test, the examined compounds were added to the culture medium, at 10 mM concentration, and incubated for 24 and 72 h incubation periods in an atmosphere of 5 % CO2 and 95 relative humidity at 37 °C.
In vitro evaluation of the cytotoxicity effects associated with the examined compounds was performed according to the protocol described in the ISO 10993-5 guide [18]. In brief, at the end of the exposure period, MTT (3-(4,5-dimethylthiazol-2yl)-2,5-diphenyl tetrazolium bromide) assay was carried out as previously described [18]. The yellow tetrazolium dye of MTT was reduced by metabolically active cells into an intracellular purple formazan product. The absorbance values of each well were determined with a microplate enzyme-linked immuno-assay (ELISA) reader equipped with a 570-nm filter. Survival rates of the controls were set to represent 100 % viability. Untreated cultures were used as control groups. Results are presented as percentage cell proliferation determined as 100- (A of experimental well/A of positive control well) X 100. Each experiment was repeated 3 times. Statistical analysis was performed applying the Student t test using SPSS 10.0 statistical software package (SPSSFW, SPSS Inc, Chicago, IL. USA) for Windows. A p value < 0.05 was considered statistically significant.
Results
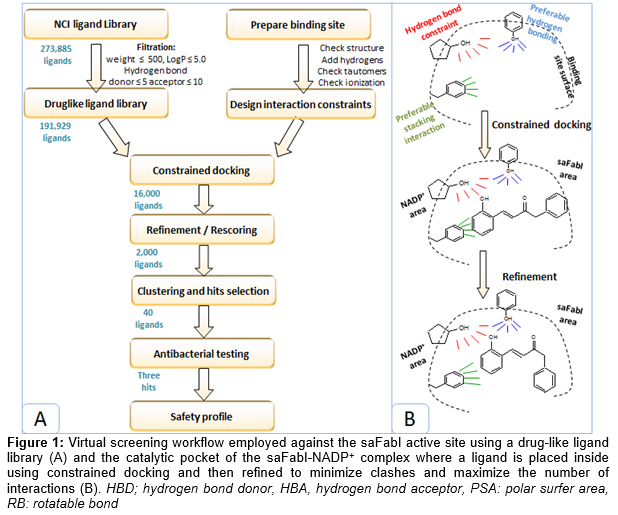
The NCI ligand library was in silico screened against the saFabI active site. The virtual screening work flow and docking methodology are schematically represented in . Prior docking, the library was filtered to satisfy the druglike rules of Lipinski and Veber, decreasing the library volume from 273,885 ligands to 191,929 ligands. Filtered ligand library was docked into the saFabI pocket where a hydrogen bonding constraint to the NADP+ ribose was applied. The top 16,000 ligands were refined and rescored in order to optimize the obtained ligand interactions and to remove any existing clashes with the surrounding residues. The top 2000 ligands were clustered and visually inspected inside the saFabI binding pocket. Consequently, 40 compounds from different chemical clusters were selected for in vitro testing.
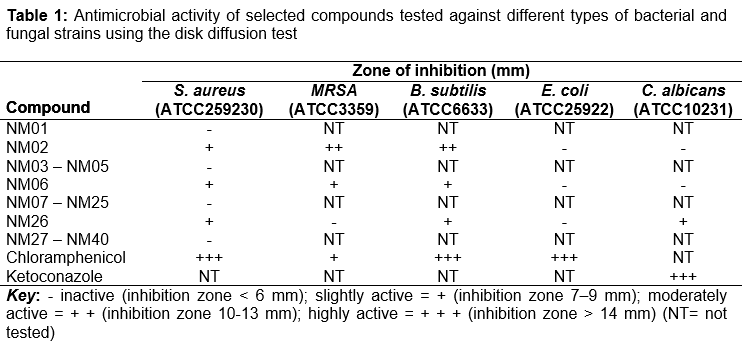
The disk diffusion test was initially used to screen the best compounds selected from virtual screening against the S. aureus strain. Among the 40 compounds tested; NM02, NM06 and NM26 showed the highest activity against S. aureus whereas the rest of the compounds revealed no antibacterial activity (). The three hits obtained from the initial in vitro screening were further tested against a wider panel of bacterial and fungal strains. Consequently, NM02 and NM06 showed varying degrees of inhibition against MRSA and B. subtilis. Whilst NM06 exhibited weak inhibition against MRSA and B. subtilis, NM02 had a moderate inhibition activity against the both gram-positive strains. In contrast, NM26 demonstrated no activity against MRSA but had slight activity against B. subtilis. None of the three hits showed inhibition activity against the gram-negative E. coli. On the other hand, NM26 showed mild inhibition activity against C. albicans while NM02 and NM06 possessed no antifungal activity.
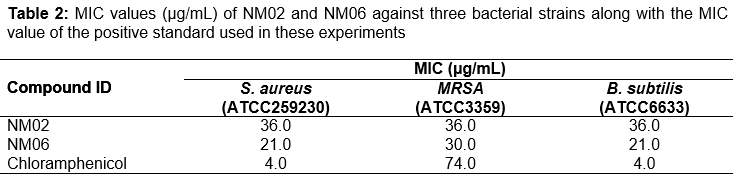
Compounds NM02 and NM06 were advanced for the next stage to identify their MIC values against the gram positive strains; results are shown in . Compound NM02 showed moderate antibacterial activity against S. aureus, MRSA and B. subtilis with a similar MIC value (36 µg/ml). Comparably, NM06 displayed moderate inhibition activity against S. aureus, MRSA and B. subtilis with MIC values of 21, 30 and 21 µg/mL respectively. Both hits had weaker activity against S. aureus and B. subtilis compared to the positive control (chloramphenicol) which had a MIC value of 4 µg/mL against the both gram-positive strains. Interestingly, NM02 and NM06 exhibited more than two-fold greater potency against MRSA compared to the reference compound chloramphenicol (MIC = 74 µg/mL).
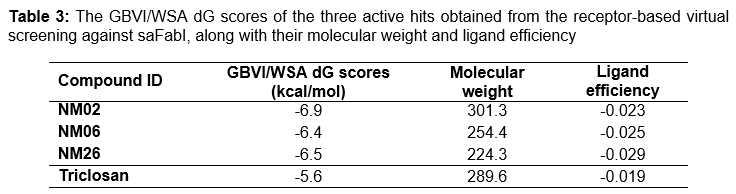
shows the names of the three discovered hits along with their predicted binding energies and molecular weight. Their binding energies were predicted to be favourable, ranging from -6.9 to -6.4 kcal/mol, scoring even better than the reference ligand triclosan (-5.6 kcal/mol). Interestingly, all three ligands showed leadlike characteristics, especially that they had molecular weight ≤ 330 da. As shown in , all three ligands were able to obtain better ligand efficiency scores (-0.023 – -0.029 kcal/mol) than the reference ligand triclosan (-0.019 kcal/mol) as they had lower binding energies and comparable or even smaller molecular weight.
On the other hand, the three active compounds were assessed for their cytotoxicity activity. After incubating the compounds with the fibroblast cell line, growth was monitored by MTT assay after 24 and 72 h. No statistically significant difference in cell proliferation was found between wells treated with the compounds under examination and the control cells (untreated well) after either 24 h or 72 h exposure periods, as shown.
Discussion
The virtual screening scheme illustrating how the drug-like ligand library was prepared, then docked into the saFabI active site is shown in A. During docking, an interaction constraint that was previously identified to enhance the ENR virtual screening [6] was also applied here; to ensure that the key interaction with NADP+ ribose will take place (B). Constrained docking was previously employed against protein tyrosine kinases [19] and phosphatases [20] and were proven to be useful in improving their virtual screening enrichment. After constrained docking,ligands were refined to relieve any existing clash that resulted from the constrained docking. Top-ranked ligands were clustered then visually inspected to finally select 40 compounds for experimental testing. Interestingly, most of the selected compounds had small sizes and contained many of the key structural features that are important for saFabI binding (e.g. hydrogen bonding with the NADP+ ribose, stacking interaction with the NADP+ nicotinamide ring and hydrogen bonding with the Tyr157 side chain).
Amongst the forty selected ligands that were screened experimentally against S. aureus, three hits turned to have antimicrobial activity (). NM02, NM06 and NM26 had also intermediate inhibitory activity against B. subtilis but with varied activities against MRSA. However, they showed no activity against the tested fungal or gram negative bacterial strains. NM02 and NM06, in particular, were further tested to identify their MIC values which indicated that to have micromolar inhibitory activity against all tested gram-positive strains (). Interestingly, these two hits had great inhibitory activity against the antibiotic resistance strain MRSA than the standard antibacterial agent, chloramphenicol. The various effects of active compounds, against the different types of microorganisms tested here, may be caused by the cell wall differences amongst the gram-positive and gram-negative strains. Additionally, the shape, size and amino acid sequence of ENRs active sites are known to have some differences, which hinder the ability of inhibitors to act as broad-spectrum antibiotics [3].
It is worth noting that NM26 is a chalcone derivative and was previously identified as an antibacterial agent. However, in the literature, there are controversial results regarding this particular compound. Kumar et al [21] found that this compound had a strong antibacterial activity against E. coli and no inhibition activity against the S. aureus strain. In contrast, Al-Mamary et al [22] found that this chalcone derivative has a strong inhibition activity against S. aureus and weak activity against E. coli along with some antifungal activity. Our findings appear more consistent with the latter finding and it suggests out that chalcones may work as antibacterial agents via inhibiting the ENR enzymes.
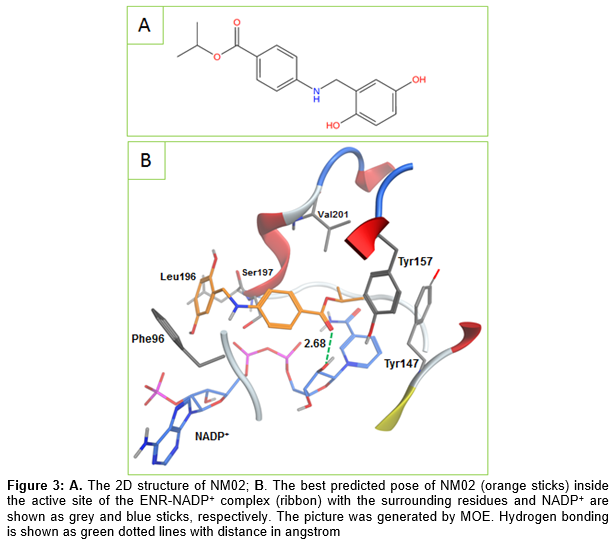
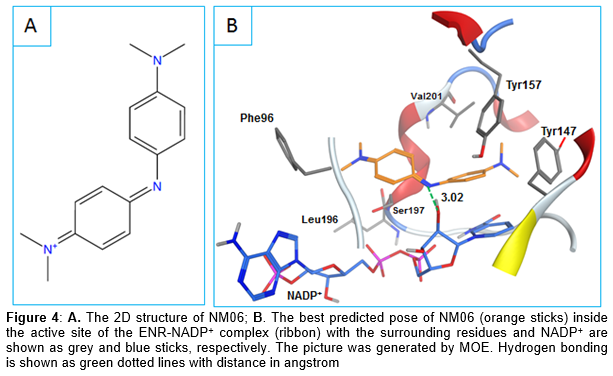
Looking at the binding modes of the three hits, it can be observed that NM02 was able to make the important hydrogen bonding with the NADP+ ribose via its carbonyl oxygen (). Additionally, it formed multiple van der Waals interactions with the surrounding residues (e.g. Leu102, Tyr147, Val201) as well as NADP+ nicotinamide ring. NM06 was also able to make the constrained interaction but this time via its imine nitrogen. As shown in , NM06 appears to be nicely filling the active site which could further stabilize its binding with the saFabI- NADP+ complex. The usual π-π interaction shown by different known ENR inhibitors was also observed here between the NM06 aromatic ring and the NADP+ nicotinamide ring.
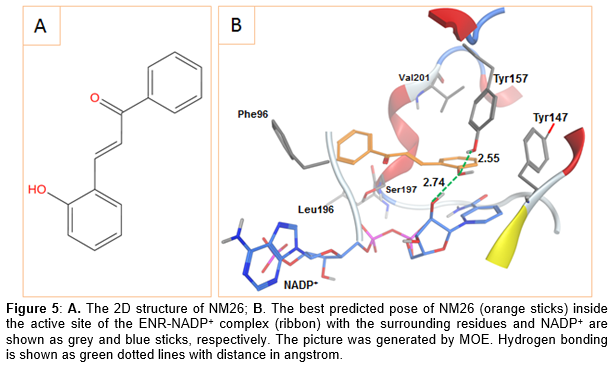
Similar to triclosan, NM26 had its hydroxyl group simultaneously interacting with the NADP+ ribose and the Tyr157 side chain via hydrogen bonding (). To sum up, the three hits seem to have what is required to interact with the ENR active site especially that all of them formed the key interaction with the NADP+ ribose hydroxyl. Additionally, two of these hits were also able to form the common hydrogen bond with Tyr157 or the usual π-π stacking interaction with the NADP+ nicotinamide ring.
In order to make use of a discovered antibacterial hit, its antimicrobial activity should be accompanied with no toxicity to the human cells. Consequently, a cytotoxicity study for the three active compounds was conducted and the three hits showed no significant toxicity to the tested human cell lines after either 24 or 72 h exposure, indicating a reasonable safety margin of the examined compounds on the normal human cells. All in all, the three hits discovered here can act as good leads to future antibacterial agents; especially as they have small structure with the important features required to bind with saFabI, and more importantly they showed experimental antimicrobial activity with minimal toxicity to human cells.
Conclusion
A validated structure-based virtual screening against the saFabI catalytic pocket has been successfully conducted. Forty hits were selected for experimental testing, three of which showed antibacterial activity, particularly, against gram-positive strains. Three compounds showed convenient docking binding modes, satisfying the key interaction with the NADP+ ribose hydroxyl group. The MIC values for the best two hits were in the micromolar range; both compounds had greater activity against MRSA compared to chloramphenicol. Interestingly, the two discovered compounds showed no toxicity towards human cells, indicating a good safety profile. The two hits seem to be promising leads since they have a good safety margin as well as a small molecule size that provides some room for improvement.
Declarations
Acknowledgement
References
Archives


News Updates